




Авторы
Чупров А. Д.
профессор, д.м.н., директор1
Пидодний Е. А.
врач-офтальмолог1
1Оренбургский филиал ФГАУ «НМИЦ »МНТК «Микрохирургия глаза» им. акад. С.Н. Федорова« Минздрава России, г. Оренбург, Российская Федерация
Автор для корреспонденции
Пидодний Екатерина Александровна; e-mail: nauka@ofmntk.ru.
Финансирование
Исследование не имело спонсорской поддержки.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Аннотация
Наследственные дистрофии сетчатки – клинически и генетически гетерогенная группа состояний, многие из которых имеют схожие симптомы и функциональные данные, что затрудняет диагностику и правильную постановку диагноза. Корнеоретинальная дистрофия Биетти – редкая форма наследственных дистрофий сетчатки с аутосомно-рецессивным типом наследования. При данной дистрофии визуализируются желто-белые кристаллы на сетчатке и наблюдается прогрессирующая атрофия пигментного эпителия сетчатки, скопление пигмента, склерозирование хориокапилляров. К развитию заболевания приводит мутация в гене CYP4V2. В настоящее время описано 50 различных мутаций CYP4V2, большинство из которых являются миссенс-мутациями. В данной статье представлен клинический случай генетически подтвержденной корнеоретинальной дистрофии Биетти с описанием клинико-функциональных признаков, на которые необходимо обратить внимание при постановке диагноза и учитывать в дифференциальной диагностике других наследственных дистрофий сетчатки. Проведение молекулярно-генетического анализа необходимо в сомнительных клинических случаях, а также для генетического консультирования родственников из группы риска и пренатального тестирования беременности с высоким риском.
Ключевые слова
корнеоретинальная дистрофия Биетти, генетический анализ, полное экзомное секвенирование, ген CYP4V2, наследственные дистрофии сетчатки, миссенс-мутации
Для цитирования
Чупров А. Д., Пидодний Е. А. Дифференциально-клинические признаки корнеоретинальной дистрофии Биетти (клинический случай). Медицина 2022; 10(1): 60-69.
DOI
Введение
Клинические симптомы, результаты тестирования поля зрения и электрофизиологических исследований корнеоретинальной дистрофии Биетти аналогичны наследственным дистрофиям сетчатки, которые попадают под категорию пигментного ретинита, что часто приводит к ошибочной постановке диагноза пациенту. Кристаллическая корнеоретинальная дистрофия Биетти – наследственная дистрофия сетчатки, впервые описанная Биетти в 1937 году [3]. Тип наследования – аутосомно-рецессивный. Заболевание характеризуется многочисленными сверкающими желто-белыми кристаллами на сетчатке и прогрессирующей атрофией пигментного эпителия сетчатки и хориокапилляров [2,3]. Маленькие блестящие кристаллы могут также встречаться в лимбе роговицы и циркулирующих лимфоцитах. Кристаллические отложения сетчатки выявляются преимущественно в заднем полюсе сетчатки в наружных и внутренних слоях сетчатки [3,8].
Корнеоретинальная дистрофия обычно проявляется к 20-40 годам, клинически характерна никталопия, снижение остроты зрения и сужение полей зрения. Данное заболевание имеет широкую распространенность в Китае и Японии. Патогенные варианты CYP4V2 были обнаружены у 97,7% лиц китайского происхождения [5,6]. Поэтому необходимо уточнять происхождение и национальность пациента.
Локус гена, ответственного за дистрофию, был картирован в 4q35-qter с помощью анализа генетического сцепления в нескольких семействах с дистрофией Биетти, и впоследствии CYP4V2 был идентифицирован как причинный ген [7,9]. Ген CYP4V2 состоит из 11 экзонов, которые кодируют широко экспрессируемый член семейства цитохрома P450 из 525 аминокислот. Цитохром Р 450 участвует в синтезе жирных кислот. В настоящее время описано 50 различных мутаций CYP4V2, большинство из которых являются миссенс-мутациями [4].
Диагноз устанавливается на основании клиническо-функциональной картины, данных генетического анализа, учитывается национальность пациента. В данной статье описан клинический случай пациентки с генетически подтвержденным диагнозом корнеоретинальной дистрофии Биетти, ранее наблюдавшейся с диагнозом тапеторетинальная абиотрофия.
Цель
Цель – представить дифференциально-клинические признаки корнеоретинальной дистрофии Биетти, отличающие данную наследственную дистрофию от тапеторетинальной абиотрофии (пигментного ретинита).
Материал и методы
Пациентке проведено комплексное клинико-функциональное обследование, включающее визометрию, кинетическую периметрию, авторефрактометрию, офтальмоскопию, гониоскопию, оптическую когерентную томографию макулярной зоны на томографе Spectralis HRA+OCT и роговицы (Орtovue RTVue-100), электроретинографию в программе ERG OU стандарт (Tomey EP-1000), сделаны снимки глазного дна в режиме multicolor, выполненные на оптическом когерентном томографе Spectralis HRA+OCT. Собран генеалогический анамнез и выполнено полное экзомное секвенирование методом NGS с последующей консультацией офтальмогенетика ФГБНУ "МГНЦ".
Результаты
Пациентка А., 51 год, обратилась в Оренбургский филиал МНТК "Микрохирургия глаза" с жалобами на снижение центрального зрения, прогрессивное сужение полей зрения, ухудшение сумеречного зрения в последние 10 лет. С 2005 года наблюдается с диагнозом тапеторетинальная абиотрофия OU. Диагноз выставлен офтальмологом по месту жительства, ежегодно получала курсы консервативной нейропротекторной, ретинопротекторной и метаболической терапии. В анамнезе в 2009 году выполнена факоэмульсификация катаракты с имплантацией интраокулярной линзы обоих глаз. Соматическая патология отсутствует.
Пациентка по национальности – китаянка. Генеалогический анамнез не отягощен ни по материнской, ни по отцовской линии. Родители клинически здоровы. Сестра пациентки не имеет клинических проявлений заболевания.
Острота зрения ОD=0,2 не корригирует, OS=0,1 не корригирует. По данным авторефрактометрии: OD – смешанный астигматизм (sph (+)0,5Dсyl (-)0,75 D), OS – смешанный астигматизм (sph (+)0,5Dсyl (-)1,0 D). При кинетической периметрии OU отмечается концентрическое сужение полей зрения до 10-30 градусов от точки фиксации (величина объекта 3 мм) (рис.1).
Рис. 1. Кинетическая периметрия
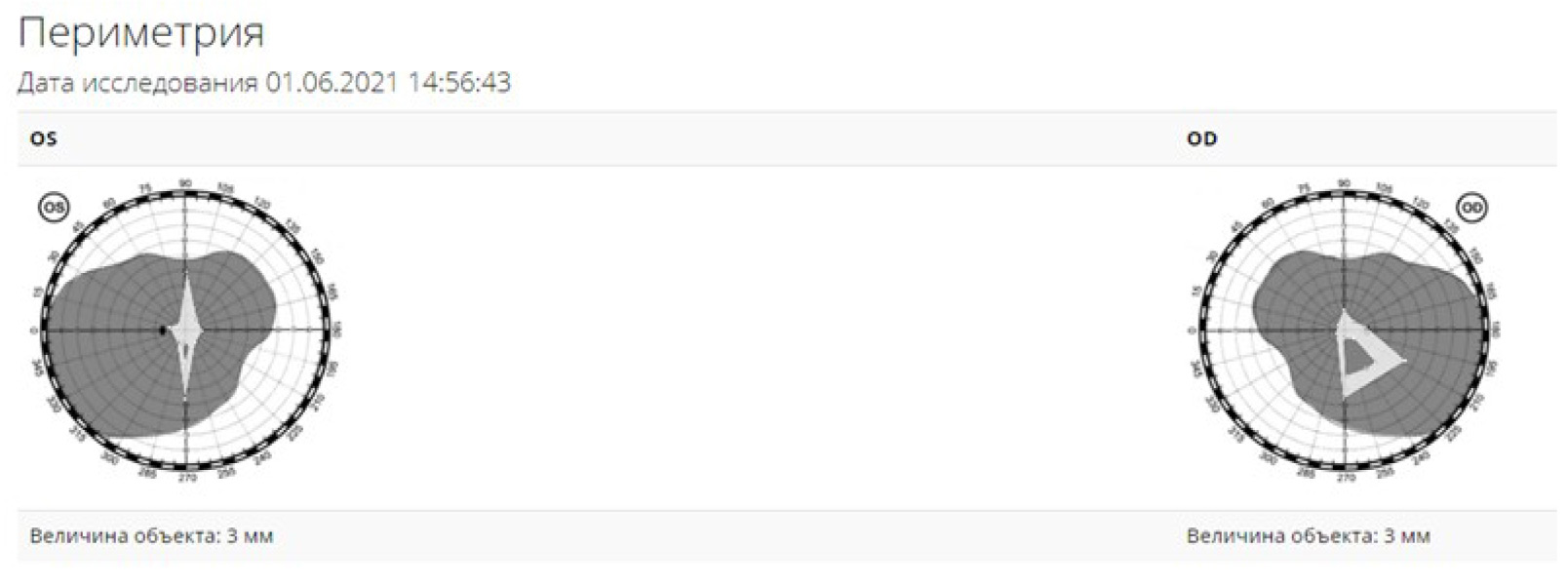
При биомикроскопии переднего отрезка OU спокойны, роговица прозрачная, передняя камера средняя, влага прозрачная, радужка спокойная, зрачок 3 мм, реакция на свет сохранена, артифакия, положение ИОЛ правильное, в стекловидном теле – нитчатая деструкция. Офтальмоскопия глазного дна: ДЗН деколорирован, монотонный, на сетчатке визуализируются множественные кристаллические отложения по заднему полюсу с максимальной концентрацией в парамакулярной зоне, отложение пигмента, перераспределение пигмента, атрофия пигментного эпителия на крайней, средней периферии, просматриваются сосуды хориоидеи. На рис. 2 представлены снимки в режиме multicolor, выполненные на оптическом когерентном томографе Spectralis HRA+OCT (Heidelberg).
Рис. 2. Снимки в режиме multicolor (Spectralis HRA+OCT Heidelberg) пациентки с корнеоретинальной дистрофией Биетти.
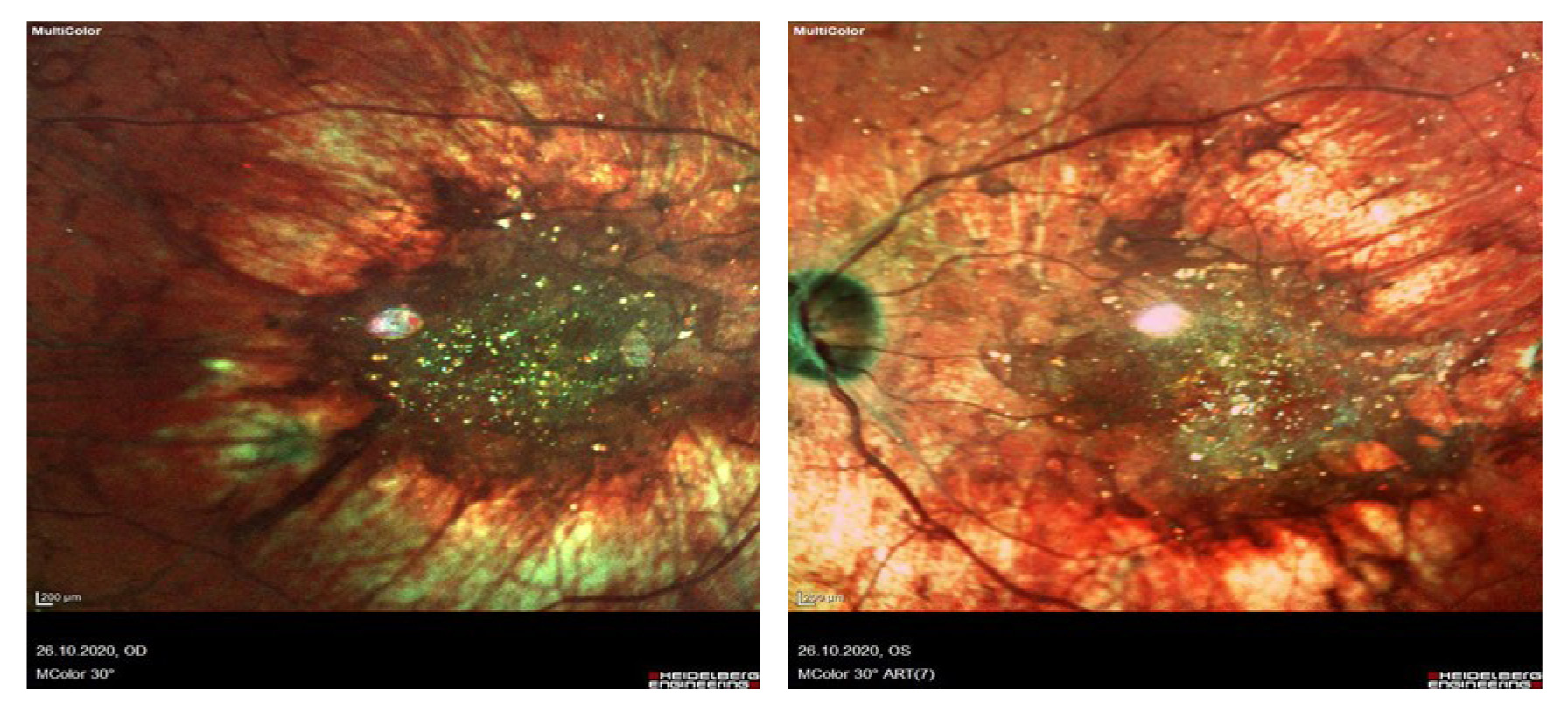
Рис. 3. Оптическая когерентная томография сетчатки spectralis HRA+OCT (Heidelberg) пациентки с корнеоретинальной дистрофией Биетти.
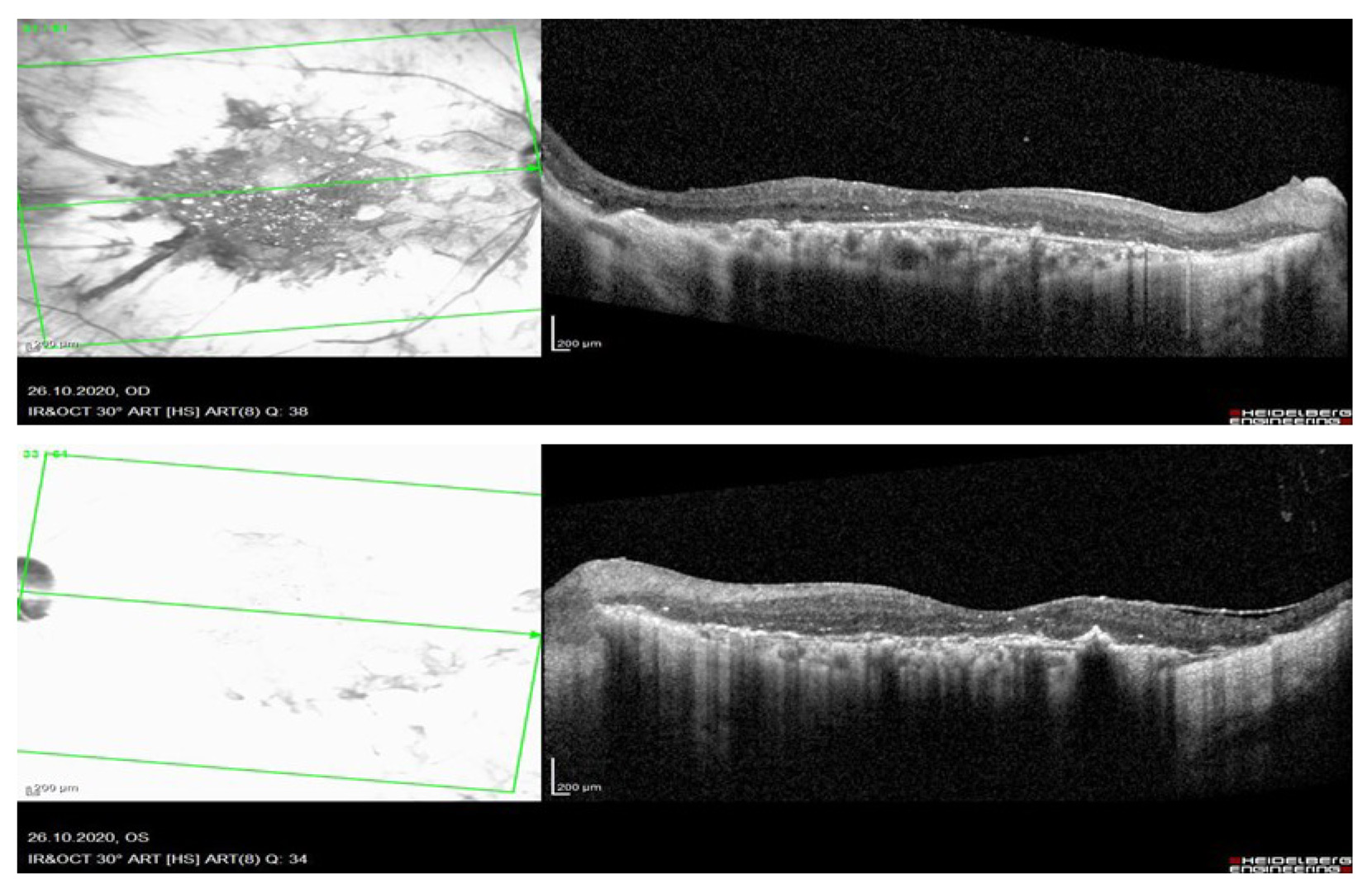
Результаты оптической когерентной томографии макулярной зоны (Spectralis HRA+OCT Heidelberg): OU Профиль сетчатки деформирован, эпиретинальный фиброз, тангенциально-тракционный синдром, мелкие гиперрефлективные очаги во всех слоях сетчатки. Участки разрушения эллипсоидной зоны и атрофия пигментного эпителия сетчатки, хориосклероз (рис.3).
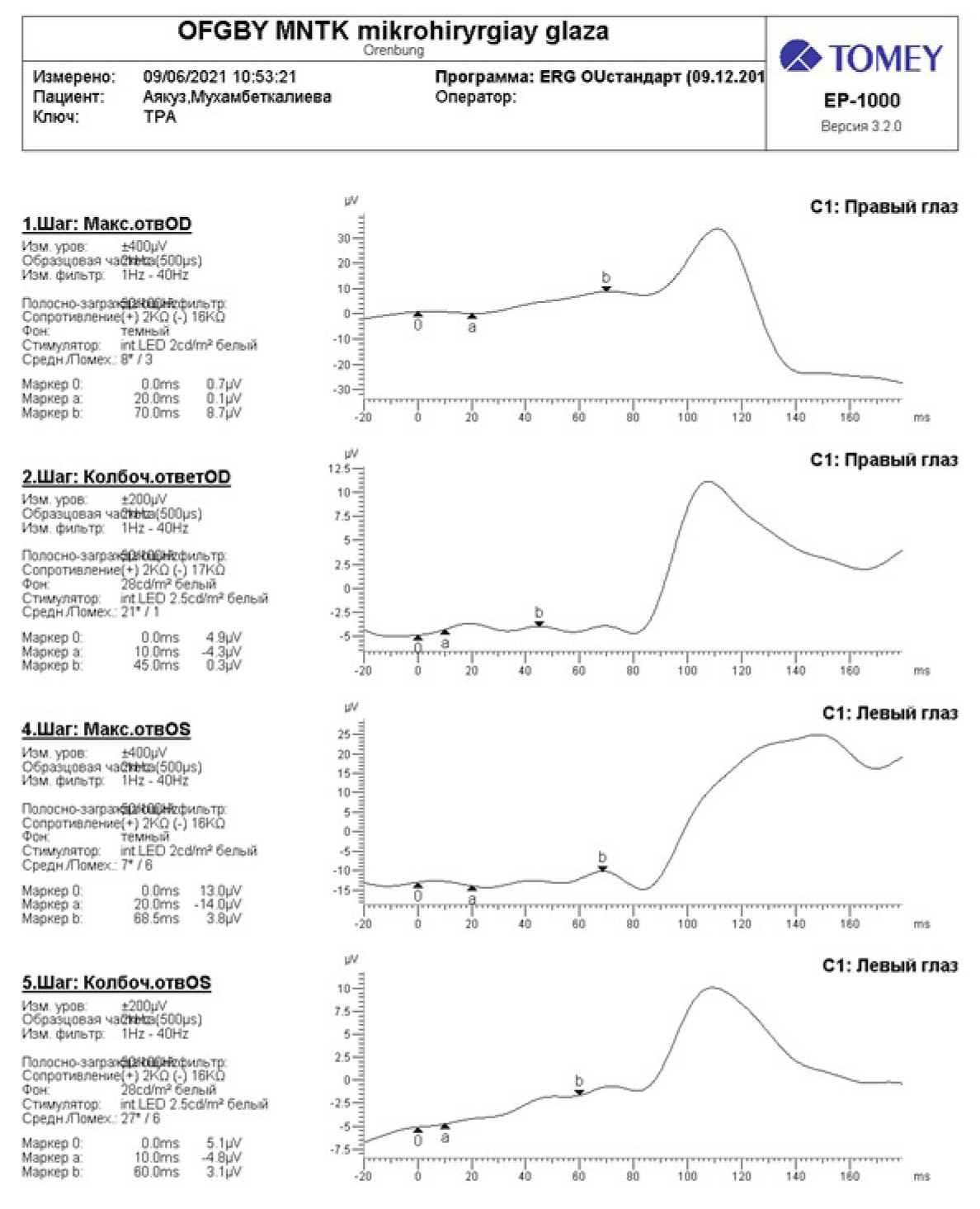
Пациентке проведена электроретинография с использованием программы ERG OU стандарт (Tomey EP-1000). По данным ЭРГ амплитуда волн "а" и "в" резко снижена при максимальном и колбочковом ответе, увеличение латентности общего биопотенциала сетчатки, что свидетельствует о поражении палочковой и колбочковой системы, регистрируется "угасающая" ЭРГ (рис. 4).
Рис. 4. Электроретинография (программа ERG OU стандарт Tomey EP-1000).
Пациентке выполнен генетический анализ – полное экзомное секвенирование методом NGS. Это генетический тест, который помогает обнаружить изменения (варианты) в генетическом коде пациента. У пациентки обнаружен ранее описанный в литературе вариант (rs1300138505) в гомозиготном состоянии в 7 экзоне (из 11) в гене CYP4V2, приводящий к нарушению канонического сайта сплайсинга (c.802-8_810delinsGC) [10,12]. Патогенные биаллельные варианты в данном гене могут приводить к развитию корнеоретинальной дистрофии Биетти. Обнаруженный вариант был описан в гомозиготном/компаунд-гетерозиготном состоянии у пациентов с корнеоретинальной дистрофией Биетти [1].
Пациентка была проконсультирована офтальмогенетиком ФГБНУ "МГНЦ". На основании полученных результатов выставлен диагноз – корнеоретинальная дистрофия тип Биетти. Рекомендовано дополнительное проведение кератотопографии, ОКТ роговицы, переднего отрезка.
После генетической верификации диагноза проведена оптическая когерентная томография роговицы на томографе Орtovue RTVue-100.
Рис. 5. Оптическая когерентная томография роговицы (Орtovue RTVue-100).
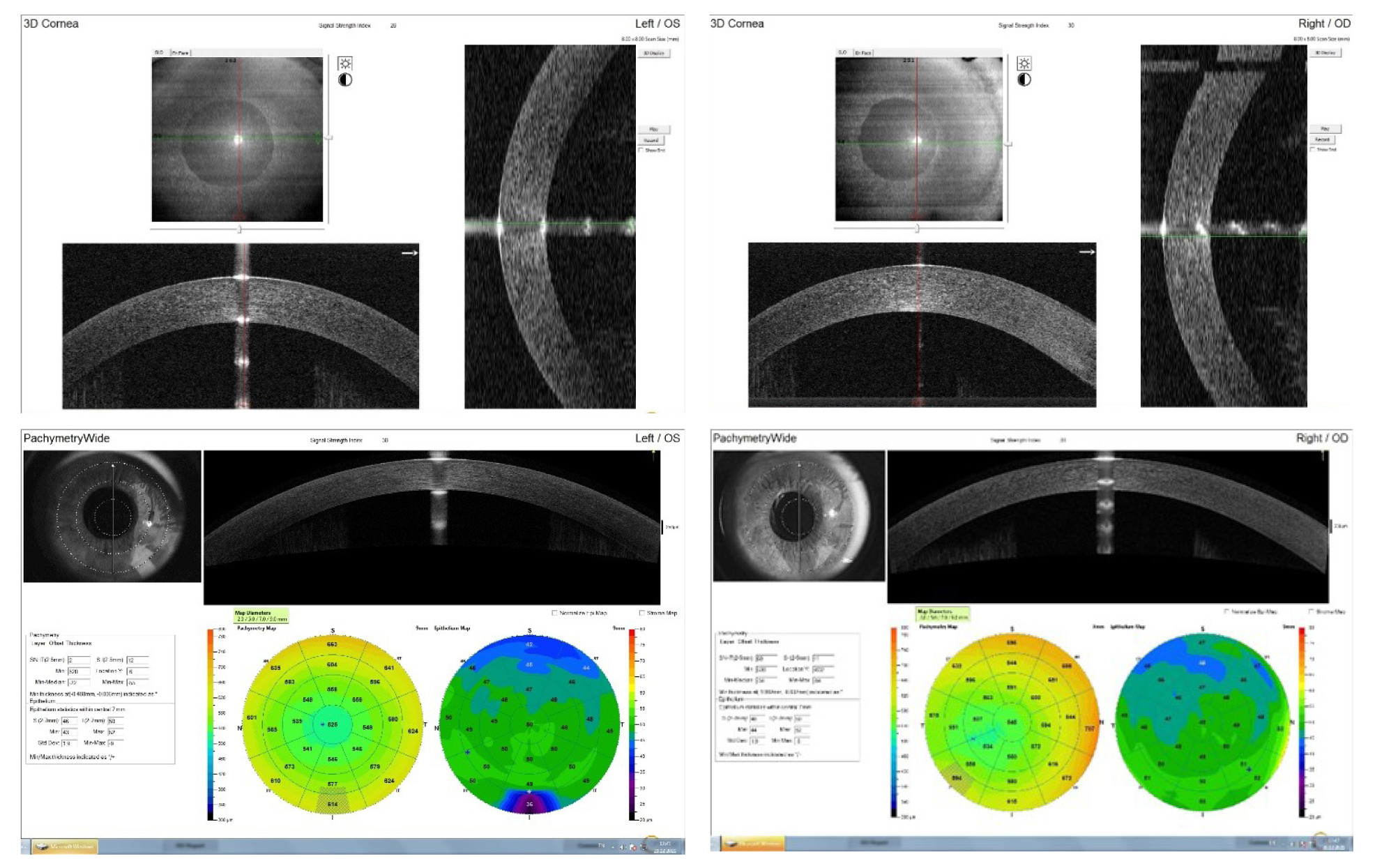
По данным оптической когерентной томографии роговицы OU определяется смещение центра роговицы, неравномерная толщина, помутнение роговицы в параоптической зоне, истончение эпителия в верхнем и нижнем отделах (на OS в большей степени) (рис. 5).
Пациентке назначена консервативная нейро-, ретинопротекторная, метаболическая и кератопластическая терапия.
Таким образом, скопление пигмента, его перераспределение на крайней и средней периферии сетчатки может ошибочно трактоваться как "костные тельца" (скопления пигмента в интраретинальных слоях), а наличие у пациента никталопии, концентрических суженных полей зрения приводит к неправильной постановке диагноза – тапеторетинальная абиотрофия (пигментный ретинит). Схожие электрофизиологические показатели ("угасающая", "нерегистрируемая" ЭРГ) могут подтвердить ошибочный диагноз. Поэтому важными клиническими признаками корнеоретинальной дистрофии Биетти является кристаллические включения на сетчатке, определяющиеся офтальмоскопически преимущественно у заднего полюса, у некоторых пациентов вариабельные кристаллические отложения обнаруживаются в лимбе роговицы [11]. На оптической когерентной томографии сетчатки кристаллические отложения визуализируются как гиперрефлективные образования во всех слоях сетчатки, причем локализация многих этих включений может не совпадать с офтальмоскопической картиной. На оптической когерентной томографии также визуализируется атрофия пигментного эпителия, разрушение эллипсоидной зоны, что характерно и для тапеторетинальной абиотрофии, но при последней эллипсоидная зона длительно сохранна в субфовеолярной зоне и имеет характерный вид (рис. 6).
Рис. 6. Оптическая когерентная томография сетчатки Spectralis HRA+OCT (Heidelberg) пациентки с PDЕ-6А, ассоциированным пигментным ретинитом.
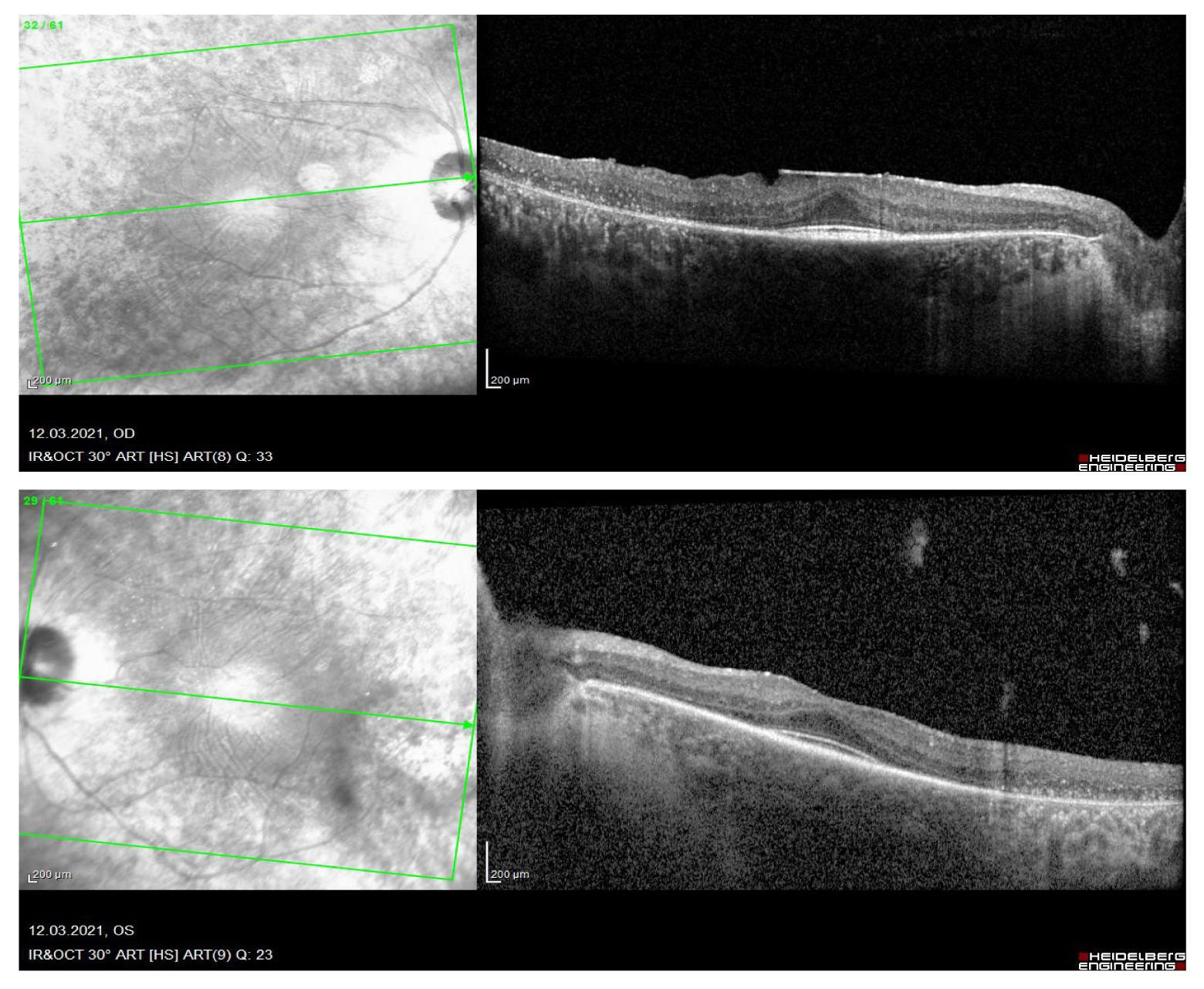
Проведение молекулярно-генетического анализа необходимо, если клинические данные не убедительны. Кроме того, выявление патогенных вариантов мутации у носителей необходимо для генетического консультирования родственников из группы риска и пренатального тестирования беременности с высоким риском.
В настоящее время таргетное лечение данной наследственной дистрофии сетчатки отсутствует. Ежегодное офтальмологическое обследование необходимо для мониторинга, оценки прогрессирования заболевания и установки инвалидности, получения средств технической реабилитации.
Знание мутации гена, которая приводит к развитию заболевания, позволяет отслеживать новые клинические исследования в сфере геномного редактирования и принимать участие в клинических исследованиях новых методов лечения.
Вывод
Корнеоретинальная дистрофия Биетти имеет характерную клиническую картину. Основными дифференциально-клиническими признаками, отличающими дистрофию Биетти от других наследственных дистрофий сетчатки, являются кристаллические отложения в сетчатке и роговице на фоне прогрессирующей атрофии пигментного эпителия и разрушения эллипсоидной зоны, манифестации заболевания к 30-40 годам, а также высокая встречаемость у лиц китайского происхождения.
Список литературы
1. Astuti G.D., Sun V., Bauwens M. et al. Novel insights into the molecular pathogenesis of CYP4V2-associated Bietti's retinal dystrophy. Mol Genet Genomic Med. 2015; 3(1): 14-29. doi: 10.1002/mgg3.109.
2. Bagolini B., Ioli-Spada G. Bietti's tapetoretinal degeneration with marginal corneal dystrophy. Am J Ophthalmol. 1968; 65(1): 53-60. doi: 10.1016/0002-9394(68)91028-3.
3. Bietti G. Ueber familiaeres Vorkommen von ‘Retinitis punctata albescens’ (verbunden mit ‘Dystrophia marginalis cristallinea corneae’), Glitzern des Glaskoerpers und anderen degenerativen Augenveraenderungen. Klin Monatsbl Au enheikd 1937; 99: 737-757.
4. Halford S., Liew G., Mackay D.S., Sergouniotis P.I., Holt R. et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology 2014; 121: 1174-84. doi: 10.1016/j.ophtha.2013.11.042.
5. Hu D.N. Genetic aspects of retinitis pigmentosa in China. Am J Med Genet. 1982; 12(1): 51–56. doi: 10.1002/ajmg.1320120107.
6. Hu D.N. Ophthalmic genetics in China. Ophthal Paediat Genet. 1983; 2: 39-45. doi: 10.3109/13816818309007148.
7. Jiao X., Munier F.L., Iwata F., Hayakawa M., Kanai A. et al. Genetic linkage of Bietti crystallin corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet. 2000; 67(5): 1309-1313. doi: 10.1016/S0002-9297(07)62960-7.
8. Kaiser-Kupfer M.I., Chan C.C., Markello T.C., Crawford M.A., Caruso R.C., Csaky K.G., Guo J., Gahl W.A. Clinical biochemical and pathologic correlations in Bietti's crystalline dystrophy. Am J Ophthalmol. 1994; 118(5): 569-582. doi: 10.1016/s0002-9394(14)76572-9.
9. Li A., Jiao X., Munier F.L., Schorderet D.F., Yao W. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004; 74(5): 817–826. doi: 10.1086/383228.
10. Park Y.J., Hwang D.J., Seong M.W., Park S.S., Woo S.J. Bietti Crystalline Retinopathy Confirmed by Mutation of CYP4V2 Gene in a Korean Patient. Korean J Ophthalmol. 2016; 30(1): 81-3. doi: 10.3341/kjo.2016.30.1.81.
11. Vargas M., Mitchell A., Yang P. et al. Bietti Crystalline Dystrophy. GeneReviews [Internet] 2012. Available at: https://www.ncbi.nlm.nih.gov/books/NBK91457/.
12. Wada Y., Itabashi T., Sato H., Kawamura M., Tada A., Tamai M. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti's crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005; 139(5): 894-9. doi: 10.1016/j.ajo.2004.11.065.
Differential Clinical Signs of Bietti Corneoretinal Dystrophy (Clinical Case)
Authors
Chuprov A. D.
Professor, Doctor of Medicine, Director1
Pidodniy E. A.
Ophthalmologist1
1Orenburg branch of the S. Fyodorov Eye Microsurgery Federal State Institution of the Ministry of Health of the Russian Federation, Orenburg, Russian Federation
Corresponding Author
Pidodniy E. A., e-mail: nauka@ofmntk.ru.
Funding
The study had no sponsorship.
Conflict of interest
None declared.
Abstract
Hereditary retinal dystrophies are clinically and genetically heterogeneous group of conditions, many of which have similar symptoms and functional findings, making correct diagnosis difficult. Bietti corneoretinal dystrophy is a rare form of hereditary retinal dystrophy with an autosomal recessive mode of inheritance. With this dystrophy, yellow-white crystals are visualized on the retina and progressive atrophy of the retinal pigment epithelium, pigment accumulation, and sclerosis of the choriocapillaries are observed. A mutation in the CYP4V2 gene leads to the development of the disease. Currently, 50 different CYP4V2 mutations have been described, most of which are missense mutations. The article presents a clinical case of genetically confirmed Bietti corneoretinal dystrophy with a description of the clinical and functional signs that should be considered when making a diagnosis and taken into account in the differential diagnosis of other hereditary retinal dystrophies. Molecular genetic analysis is necessary in doubtful clinical cases, as well as for genetic counseling of relatives at risk and prenatal testing of high-risk pregnancies.
Key words
Bietti corneoretinal dystrophy, genetic analysis, whole exome sequencing, CYP4V2 gene, hereditary retinal dystrophies, missense mutations
DOI
References
1. Astuti G.D., Sun V., Bauwens M. et al. Novel insights into the molecular pathogenesis of CYP4V2-associated Bietti's retinal dystrophy. Mol Genet Genomic Med. 2015; 3(1): 14-29. doi: 10.1002/mgg3.109.
2. Bagolini B., Ioli-Spada G. Bietti's tapetoretinal degeneration with marginal corneal dystrophy. Am J Ophthalmol. 1968; 65(1): 53-60. doi: 10.1016/0002-9394(68)91028-3.
3. Bietti G. Ueber familiaeres Vorkommen von ‘Retinitis punctata albescens’ (verbunden mit ‘Dystrophia marginalis cristallinea corneae’), Glitzern des Glaskoerpers und anderen degenerativen Augenveraenderungen. Klin Monatsbl Au enheikd 1937; 99: 737-757.
4. Halford S., Liew G., Mackay D.S., Sergouniotis P.I., Holt R. et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology 2014; 121: 1174-84. doi: 10.1016/j.ophtha.2013.11.042.
5. Hu D.N. Genetic aspects of retinitis pigmentosa in China. Am J Med Genet. 1982; 12(1): 51–56. doi: 10.1002/ajmg.1320120107.
6. Hu D.N. Ophthalmic genetics in China. Ophthal Paediat Genet. 1983; 2: 39-45. doi: 10.3109/13816818309007148.
7. Jiao X., Munier F.L., Iwata F., Hayakawa M., Kanai A. et al. Genetic linkage of Bietti crystallin corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet. 2000; 67(5): 1309-1313. doi: 10.1016/S0002-9297(07)62960-7.
8. Kaiser-Kupfer M.I., Chan C.C., Markello T.C., Crawford M.A., Caruso R.C., Csaky K.G., Guo J., Gahl W.A. Clinical biochemical and pathologic correlations in Bietti's crystalline dystrophy. Am J Ophthalmol. 1994; 118(5): 569-582. doi: 10.1016/s0002-9394(14)76572-9.
9. Li A., Jiao X., Munier F.L., Schorderet D.F., Yao W. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004; 74(5): 817–826. doi: 10.1086/383228.
10. Park Y.J., Hwang D.J., Seong M.W., Park S.S., Woo S.J. Bietti Crystalline Retinopathy Confirmed by Mutation of CYP4V2 Gene in a Korean Patient. Korean J Ophthalmol. 2016; 30(1): 81-3. doi: 10.3341/kjo.2016.30.1.81.
11. Vargas M., Mitchell A., Yang P. et al. Bietti Crystalline Dystrophy. GeneReviews [Internet] 2012. Available at: https://www.ncbi.nlm.nih.gov/books/NBK91457/.
12. Wada Y., Itabashi T., Sato H., Kawamura M., Tada A., Tamai M. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti's crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005; 139(5): 894-9. doi: 10.1016/j.ajo.2004.11.065.

